 Der Begriff Mukoviszidose, auch zystische Fibrose genannt, kommt aus dem Lateinischen und setzt sich zusammen aus mucus für Schleim und viscidus für klebrig oder zäh. Bei der autosomal-rezessiv vererbten Stoffwechselerkrankung bildet sich in Drüsenzellen durch eine Mutation im CFTR-Gen vermehrt viskoser und zäher Schleim, der in den betroffenen Organen zu Funktionsstörungen führt.
Der Begriff Mukoviszidose, auch zystische Fibrose genannt, kommt aus dem Lateinischen und setzt sich zusammen aus mucus für Schleim und viscidus für klebrig oder zäh. Bei der autosomal-rezessiv vererbten Stoffwechselerkrankung bildet sich in Drüsenzellen durch eine Mutation im CFTR-Gen vermehrt viskoser und zäher Schleim, der in den betroffenen Organen zu Funktionsstörungen führt.
Autosomal-rezessive Vererbung
Diploide Organismen haben von jedem Gen zwei Kopien – eins von der Mutter und eins vom Vater. Ein homozygotes Erbgut liegt vor, wenn beide Kopien (Allele) identisch sind, ein heterozygotes, wenn sie sich unterscheiden. Dabei setzt sich immer eine Variante durch, die den Phänotypen bestimmt. Dieses Allel ist dominant gegenüber dem anderen. Dieses liegt rezessiv vor und tritt nicht in Erscheinung. 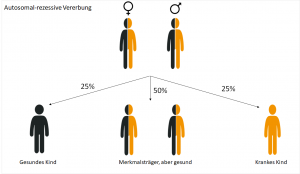 Bei Krankheiten, die autosomal-rezessiv vererbt werden, liegt die genetische Ursache auf einen der 22 Autosomen. Die Eltern tragen das veränderte Merkmal, das jedoch nicht zum Vorschein kommt. Sie sind somit gesund. Ein Kind erkrankt an Mukoviszidose, wenn es jeweils von beiden Elternteilen das veränderte Allel vererbt bekommt. Dies geschieht mit einer Wahrscheinlichkeit von 25%. Kinder, die jeweils ein wildtypisches und ein krankes Allel vererbt bekommen, sind gesund, aber wie ihre Eltern, Merkmalsträger. Sie leiden zwar nicht an der Krankheit, können diese aber an die nachfolgende Generation weitergeben.
Bei Krankheiten, die autosomal-rezessiv vererbt werden, liegt die genetische Ursache auf einen der 22 Autosomen. Die Eltern tragen das veränderte Merkmal, das jedoch nicht zum Vorschein kommt. Sie sind somit gesund. Ein Kind erkrankt an Mukoviszidose, wenn es jeweils von beiden Elternteilen das veränderte Allel vererbt bekommt. Dies geschieht mit einer Wahrscheinlichkeit von 25%. Kinder, die jeweils ein wildtypisches und ein krankes Allel vererbt bekommen, sind gesund, aber wie ihre Eltern, Merkmalsträger. Sie leiden zwar nicht an der Krankheit, können diese aber an die nachfolgende Generation weitergeben.
Ionenkanäle
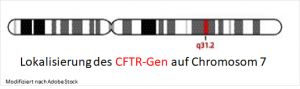
1938 hat eine amerikanische Kinderärztin die Krankheit erstmals beschrieben, knapp 50 Jahre danach wurde das das dafür verantwortliche Gen entdeckt:1 CFTR (cystic fibrosis transmembrane conductance regulator) besteht aus etwa 6.500 Basenpaare und befindet sich auf den langen Arm des Chromosom 7.2 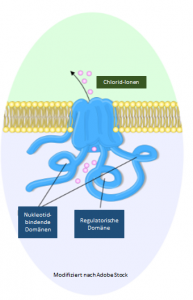 Es enthält die Bauanleitung für einen Kanal in der Zellmembran, über den Ionen in und aus der Zelle gelangen.3 Solche Kanäle, auch Ionenkanäle genannt, sind röhrenförmig angeordnete Proteinkomplexe, die in der Membran eingelagert und selektiv für bestimmte Ionen sind.4 Das Gen CFTR codiert einen Chloridkanal, der hauptsächlich in Epithelien von Darm, Bauchspeicheldrüse, Schweißdrüse und Luftwegen vorkommt. Das 1.440 Aminosäuren lange Transmembranprotein besteht aus einer regulatorischen Untereinheit mit mehreren Phosphorylierungsstellen und zwei Nukleotid-bindenden Domänen, die ATP zu ADP hydrolysieren.3 Die Hauptaufgabe von CFTR ist die Regulation des Salz- und Wasserhaushaltes menschlicher Zellen.3
Es enthält die Bauanleitung für einen Kanal in der Zellmembran, über den Ionen in und aus der Zelle gelangen.3 Solche Kanäle, auch Ionenkanäle genannt, sind röhrenförmig angeordnete Proteinkomplexe, die in der Membran eingelagert und selektiv für bestimmte Ionen sind.4 Das Gen CFTR codiert einen Chloridkanal, der hauptsächlich in Epithelien von Darm, Bauchspeicheldrüse, Schweißdrüse und Luftwegen vorkommt. Das 1.440 Aminosäuren lange Transmembranprotein besteht aus einer regulatorischen Untereinheit mit mehreren Phosphorylierungsstellen und zwei Nukleotid-bindenden Domänen, die ATP zu ADP hydrolysieren.3 Die Hauptaufgabe von CFTR ist die Regulation des Salz- und Wasserhaushaltes menschlicher Zellen.3
Mutationen im CFTR-Gen
Die Ursache der Stoffwechselerkrankung Mukoviszidose liegt bei den CFTR-Proteinen. Dadurch wird der Ionen-Transport blockiert und es herrscht ein Ungleichgewicht im Salz- und Wasserhaushalt der Zellen. Diese sind nicht mehr in der Lage mittels Osmose Wasser in die umliegenden Gewebe zu ziehen, was zur Folge hat, dass sich in Lunge, Nasennebenhöhlen, Bauchspeicheldrüse, Darm, Gallenwege sowie Keimdrüsen Schleimsekrete mit zu hohem Salzgehalt bilden. Durch den osmotischen Wasserentzug ist der Wassergehalt in den entsprechenden Sekreten zu niedrig – diese werden zähflüssig und führen in den betroffenen Organen zu Funktionsstörungen.2 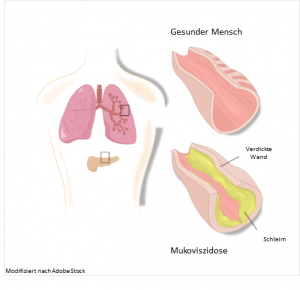 Bisher sind knapp 2000 Mutationen bekannt, die sich in sechs Klassen unterteilen lassen:5,6
Bisher sind knapp 2000 Mutationen bekannt, die sich in sechs Klassen unterteilen lassen:5,6 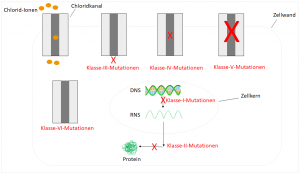 Mutationen der Klasse I betreffen ca. 10% aller Patienten. Diese entstehen durch eine defekte mRNS, sodass das entsprechende Protein nicht gebildet wird. Bei Menschen mit Mutationen der Klasse II wird zwar die RNS transkribiert, jedoch führt eine Mutation zu einem fehlerhaften Protein, welches daraufhin enzymatisch abgebaut wird. Die häufigste vorkommende Mutation in Deutschland, die bei 70% der Patienten auftritt, ist eine Deletion von drei Basenpaaren. Die entsprechende Aminosäure wird nicht gebildet, was zu einem funktionslosen Protein führt. Bei Mutationen der Klasse III liegt eine Regulationsstörung vor, sodass der Kanal nicht geöffnet wird. Im Gegensatz zur Klasse IV, bei der eine verminderte Leitfähigkeit vorliegt, was zur Folge hat, dass die Ionen die Fähigkeit verlieren, den Kanal zu passieren. Folgen von Klasse V und VI Mutationen sind zu wenige Kanäle oder instabile, die vor Erreichen der Zellmembran bereits abgebaut werden.5,6
Mutationen der Klasse I betreffen ca. 10% aller Patienten. Diese entstehen durch eine defekte mRNS, sodass das entsprechende Protein nicht gebildet wird. Bei Menschen mit Mutationen der Klasse II wird zwar die RNS transkribiert, jedoch führt eine Mutation zu einem fehlerhaften Protein, welches daraufhin enzymatisch abgebaut wird. Die häufigste vorkommende Mutation in Deutschland, die bei 70% der Patienten auftritt, ist eine Deletion von drei Basenpaaren. Die entsprechende Aminosäure wird nicht gebildet, was zu einem funktionslosen Protein führt. Bei Mutationen der Klasse III liegt eine Regulationsstörung vor, sodass der Kanal nicht geöffnet wird. Im Gegensatz zur Klasse IV, bei der eine verminderte Leitfähigkeit vorliegt, was zur Folge hat, dass die Ionen die Fähigkeit verlieren, den Kanal zu passieren. Folgen von Klasse V und VI Mutationen sind zu wenige Kanäle oder instabile, die vor Erreichen der Zellmembran bereits abgebaut werden.5,6
Erkrankungen
Mukoviszidose ist durch die Vielzahl der unterschiedlichen Mutationen eine Multisystemerkrankung. Da der zähe Schleim vor allem die kleinen Äste der Bronchien und die Ausführungsgänge der inneren Organe verstopft, sind Atmung und Verdauung besonders stark betroffen.2 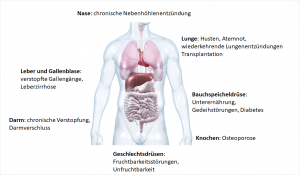 In den Bronchien ist der Ionenkanal vor allem dafür zuständig, durch Abgabe der Chlorid-Ionen den Flüssigkeitsfilm auf der Oberfläche zu erhalten. In diesem bewegen sich kleine Zilien (Flimmerhärchen), die Schmutzpartikel oder Bakterien aus der Lunge befördern. Ist dieser Prozess durch die vermehrte Schleimbildung gestört, können sich Bakterien leichter ansiedeln und Infektionen verursachen.7 Die Erkrankung der Atemwege ist auch die häufigste Todesursache bei Mukoviszidose-Patienten.1 Die Bauchspeicheldrüse gibt bei gesunden Menschen Verdauungsenzyme in den Zwölffingerdarm ab. Dort spalten diese Eiweiße, Kohlenhydrate und Fette der Nahrung. Bei Patienten mit Mukoviszidose staut sich dies als zähflüssiges Sekret an. Dies führt zu Entzündungen, der Pankreas verhärtet und vernarbt sich (Fibrose). Die Konsequenzen sind Unterernährung und Wachstumsstörungen.2 Diese Patienten haben auch ein erhöhtes Risiko an Diabetes zu erkranken (Diabetes Typ III: cystic fibrosis related diabetes, CFRD).8 Eine weitere Folge ist die Unfruchtbarkeit. 98% der Männer bilden zwar befruchtungsfähige Spermien, diese können aufgrund des Schleims den Samenleiter nicht passieren. Frauen sind ebenfalls nur bedingt fruchtbar, da der Schleim in dem Eileiter die Spermien nicht durchdringen lässt.2,8 Allen gemeinsam ist die abnehmende Lungenfunktion, sodass oftmals eine Transplantation der letzte Ausweg ist.1
In den Bronchien ist der Ionenkanal vor allem dafür zuständig, durch Abgabe der Chlorid-Ionen den Flüssigkeitsfilm auf der Oberfläche zu erhalten. In diesem bewegen sich kleine Zilien (Flimmerhärchen), die Schmutzpartikel oder Bakterien aus der Lunge befördern. Ist dieser Prozess durch die vermehrte Schleimbildung gestört, können sich Bakterien leichter ansiedeln und Infektionen verursachen.7 Die Erkrankung der Atemwege ist auch die häufigste Todesursache bei Mukoviszidose-Patienten.1 Die Bauchspeicheldrüse gibt bei gesunden Menschen Verdauungsenzyme in den Zwölffingerdarm ab. Dort spalten diese Eiweiße, Kohlenhydrate und Fette der Nahrung. Bei Patienten mit Mukoviszidose staut sich dies als zähflüssiges Sekret an. Dies führt zu Entzündungen, der Pankreas verhärtet und vernarbt sich (Fibrose). Die Konsequenzen sind Unterernährung und Wachstumsstörungen.2 Diese Patienten haben auch ein erhöhtes Risiko an Diabetes zu erkranken (Diabetes Typ III: cystic fibrosis related diabetes, CFRD).8 Eine weitere Folge ist die Unfruchtbarkeit. 98% der Männer bilden zwar befruchtungsfähige Spermien, diese können aufgrund des Schleims den Samenleiter nicht passieren. Frauen sind ebenfalls nur bedingt fruchtbar, da der Schleim in dem Eileiter die Spermien nicht durchdringen lässt.2,8 Allen gemeinsam ist die abnehmende Lungenfunktion, sodass oftmals eine Transplantation der letzte Ausweg ist.1
Epidemiologie
Mukoviszidose ist eine seltene Krankheit mit einer Prävalenz von 1/3.300 – 1/4.800 Neugeborenen in Deutschland. Dennoch ist es die zweithäufigste hereditäre Stoffwechselerkrankung der europäischen Bevölkerung (nach Hämochromatose). In Deutschland sind aktuell 8.000 Menschen davon betroffen, mit ca. 200 Neuerkrankungen pro Jahr (Stand 2017).5 Zwischen 2012 und 2016 starben 423 Menschen aufgrund von Mukoviszidose,9 im Jahr 2017 waren es 48 Todesfälle.10 Die Heterozygoten-Frequenz, also der Anteil aller Menschen, die ein krankes Allel tragen, beträgt in Europa 4% (1:25).5
Aktuelle Therapien
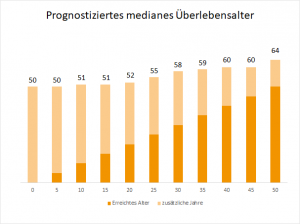 Auch wenn die Erkrankung noch nicht heilbar ist, ist die Lebenserwartung in den letzten Jahren gestiegen.9 Wurden Kinder damals nicht mal ein Jahr alt,1 liegt das prognostizierte mediane Überlebensalter eines Neugeborenen mittlerweile bei 50 Jahren.10 Aktuell sind mehr als 50% der Patienten in Deutschland älter als 18 Jahre.5 Das liegt unter anderem daran, dass es für etwa ein Drittel der Betroffenen mutationsspezifische Therapien gibt.1 Durch Genanalysen wird hier die genaue Ursache der Krankheit festgestellt. Seit 2012 stehen sogenannte CFTR-Modulatoren zur Verfügung, die ein defektes oder fehlendes Protein verbessern oder wiederherstellen.1 Die europäische Kommission hat 2012 den Wirkstoff Ivacaftor zugelassen, der die Auswirkungen des Gendefekts mildert.11 Der CFTR-Potentiator kommt für ca. 1500 Patienten in Europa in Frage, die eine Mutation der Klasse III aufweisen.12 Hier befindet sich das Protein zwar in der Zellmembran, der Kanal öffnet sich jedoch nicht. Ivacaftor stellt diese Funktion wieder her und der Transport von Chlorid-Ionen durch den Kanal wird erhöht. Grundlage für die Zulassung in Europa waren Ergebnisse einer Phase III Studie, bei sich die Lungenfunktion der Patienten signifikant verbesserte.12 Lumacaftor ist ein Wirkstoff aus der Gruppe der CFTR-Korrektoren und wird bei Mutationen der Klasse II zusammen mit Ivacaftor eingesetzt. Das fehlgefaltete Protein wird vor dem intrazellulären, enzymatischen Abbau geschützt und in der Zellmembran eingebaut. Dieses Jahr wurde ein weiterer Wirkstoff, Tezacaftor, zugelassen, der ähnlich wie Lumcaftor wirkt und ebenfalls in Kombination mit Ivacaftor eingesetzt wird.13 Studien zeigten, dass sich der Verlauf der Krankheit günstig beeinflusste, indem sich zum Beispiel die Lungenfunktion verbesserte.14,15 Ein weiterer Aspekt ist die Durchführung von sogenannten Neugeborenen-Screenings. Vor zwei Jahren wurden bundesweit Untersuchungen von Neugeborenen auf Mukoviszidose eingeführt, die kostenlos und freiwillig sind. In Bayern wurden im Zeitraum September 2016 – August 2018 insgesamt 250.000 Kinder untersucht. Bei 45 wurde die angeborene Erkrankung diagnostiziert.16 Dies ist wichtig, um mit einer frühzeitigen Therapie zu beginnen.
Auch wenn die Erkrankung noch nicht heilbar ist, ist die Lebenserwartung in den letzten Jahren gestiegen.9 Wurden Kinder damals nicht mal ein Jahr alt,1 liegt das prognostizierte mediane Überlebensalter eines Neugeborenen mittlerweile bei 50 Jahren.10 Aktuell sind mehr als 50% der Patienten in Deutschland älter als 18 Jahre.5 Das liegt unter anderem daran, dass es für etwa ein Drittel der Betroffenen mutationsspezifische Therapien gibt.1 Durch Genanalysen wird hier die genaue Ursache der Krankheit festgestellt. Seit 2012 stehen sogenannte CFTR-Modulatoren zur Verfügung, die ein defektes oder fehlendes Protein verbessern oder wiederherstellen.1 Die europäische Kommission hat 2012 den Wirkstoff Ivacaftor zugelassen, der die Auswirkungen des Gendefekts mildert.11 Der CFTR-Potentiator kommt für ca. 1500 Patienten in Europa in Frage, die eine Mutation der Klasse III aufweisen.12 Hier befindet sich das Protein zwar in der Zellmembran, der Kanal öffnet sich jedoch nicht. Ivacaftor stellt diese Funktion wieder her und der Transport von Chlorid-Ionen durch den Kanal wird erhöht. Grundlage für die Zulassung in Europa waren Ergebnisse einer Phase III Studie, bei sich die Lungenfunktion der Patienten signifikant verbesserte.12 Lumacaftor ist ein Wirkstoff aus der Gruppe der CFTR-Korrektoren und wird bei Mutationen der Klasse II zusammen mit Ivacaftor eingesetzt. Das fehlgefaltete Protein wird vor dem intrazellulären, enzymatischen Abbau geschützt und in der Zellmembran eingebaut. Dieses Jahr wurde ein weiterer Wirkstoff, Tezacaftor, zugelassen, der ähnlich wie Lumcaftor wirkt und ebenfalls in Kombination mit Ivacaftor eingesetzt wird.13 Studien zeigten, dass sich der Verlauf der Krankheit günstig beeinflusste, indem sich zum Beispiel die Lungenfunktion verbesserte.14,15 Ein weiterer Aspekt ist die Durchführung von sogenannten Neugeborenen-Screenings. Vor zwei Jahren wurden bundesweit Untersuchungen von Neugeborenen auf Mukoviszidose eingeführt, die kostenlos und freiwillig sind. In Bayern wurden im Zeitraum September 2016 – August 2018 insgesamt 250.000 Kinder untersucht. Bei 45 wurde die angeborene Erkrankung diagnostiziert.16 Dies ist wichtig, um mit einer frühzeitigen Therapie zu beginnen.
Ausblick
Auch wenn die Stoffwechselkrankheit aktuell noch nicht heilbar ist, stehen die Chancen durch CRISPR/Cas relativ gut. Dadurch lässt sich das krankhafte Gen ausschneiden und durch ein neues ersetzen, bzw. die vorliegende Mutation reparieren.1 Ein kürzlich neu entdeckter Zelltyp könnte ebenfalls einen Ansatz für zukünftige Therapien darstellen: Forscher haben diesen in der Atemschleimhaut entdeckt und Ionozyt genannt. Dieser scheint Hauptproduzent der CFTR-Proteine zu sein und kontrolliert durch das Gen FOX1 die Expression der Chloridkanäle.17,18 Somit könnte eine Stimulierung für die Neubildung der Ionozyten zu einer erfolgreichen Therapie verhelfen.19,20 Mit diesen Behandlungsoptionen wird erneut ein Fortschritt in Richtung Präzisionsmedizin geleistet. Insgesamt sind bereits 62 Wirkstoffe für die personalisierte Therapie zugelassen.21 Ansprechpartner: Kristina Schraml (kristina.schraml@biovariance.com) Quellen

