Das gemeinsame Ziel – Personalisierte Medizin voranbringen
Die BioVariance GmbH arbeitet mit einem Global Player der Pharmaindustrie zusammen, um die Forschung zur Personalisierten Medizin voranzutreiben. Dank der ausgezeichneten Zusammenarbeit und der hervorragenden Expertise beider Parteien wird dem Pharma- und Gesundheitssektor nun eine neue Kategorisierung von Krebsindikationen zur Verfügung stehen. Die neue Kategorisierung basierend auf den individuellen Charakteristika des Tumorgewebes soll ein tieferes Verständnis der Krankheitsmerkmale erzeugen sowie eine individuelle Diagnose und zukünftig eine gezieltere und personalisierte Therapie für betroffene Patienten ermöglichen.
Die Herausforderungen
Die rasante Weiterentwicklung der Messtechnologien in der biomedizinischen und pharmazeutischen Forschung ermöglicht die Generierung von komplexen interdisziplinären Datensätzen. Im Rahmen dieses Projektes verwendete der Global Player NGS (Next Generation Sequencing) und innovative optische Messmethoden, um Gewebsschnitte bestimmter Tumore zu charakterisieren. Für die Präprozessierung der Datensätze sowie für die Entwicklung und Beurteilung der Clustering Modelle beauftragte der Global Player die BioVariance GmbH aufgrund Ihrer breiten Expertise zu den Themen Datenprozessierung, explorative Statistik und Maschinelles Lernen von und mit Big Data. Um sowohl genetische als auch gewebsspezifische Daten für die Analyse bezüglich der Kategorisierung einer Krebsindikation verwenden zu können, führte die BioVariance GmbH für jeden Datensatz eine geeignete Normierung durch. In diesem Fall sollte die Normierung nicht nur die Daten vergleichbar machen, sondern zudem einen durch die neue Messtechnik möglicherweise entstandenen Bias beheben. Nach der Festlegung einer geeigneten Normierungsstrategie wurde mittels des Varianzkoeffizienten[1] sowie mithilfe partieller Korrelationen sichergestellt, dass es sich bei den in der Scatterplot-Matrix ermittelten linearen Korrelationen der betrachteten Eigenschaften um keine durch die Normierung verursachten Scheinkorrelationen handelt. Da die Scatterplot-Matrix, anders als im Beispieldatensatz der Abb. 1, keine Cluster anhand einzelner Eigenschaftsvergleiche sichtbar machte, entschied die BioVariance GmbH, im nächsten Schritt eine Hauptkomponentenanalyse durchzuführen. Dafür wurden die normierten Gewebscharakteristika zunächst einer Box-Cox-Transformation unterzogen, da keine dieser Variablen einer Normalverteilung folgte.
Das Vorgehen
Im Rahmen dieses Projektes setzten die Datenanalysten der BioVariance GmbH auf die modernsten Algorithmen der deskriptiven Statistik und der explorativen Datenanalyse, um Unterschiede, Korrelationen und Cluster in den Datensätzen aufzudecken. In dem hier beschriebenen Projekt waren keine Cluster in den Daten anhand der Scatterplots erkennbar, sodass die BioVariance GmbH eine Hauptkomponentenanalyse (PCA), inklusive der Bestimmung der strukturgebenden Faktoren durch die Marchenko-Pastur-Verteilung als nächsten Schritt wählte. Die PCA reduziert die hochdimensionalen Ausgangsdaten auf wenige Dimensionen und sucht dabei nach linearen Kombinationen der betrachteten Eigenschaften, den sogenannten Hauptkomponenten, die die größte Streuung in den Daten beschreiben und damit das größte Potential haben, Cluster in den Daten erkennbar zu machen. Die erste Hauptkomponente beschreibt dann den größten Anteil der Streuung in den Daten, die zweite den zweitgrößten, etc. Die Hauptkomponenten stellen dabei ein orthogonales Koordinatensystem dar, dessen Achsen in Richtung der größten Streuung der Daten weisen. Mit diesen lässt sich eine reduzierte Darstellung der Daten in zwei oder drei Dimensionen realisieren. Oftmals sind bereits in dieser Darstellung der Daten Cluster zu erkennen.[2] Über die Marchenko-Pastur-Verteilung wurde die Anzahl an Hauptkomponenten ermittelt, die Information tragen und damit das Potential haben, Cluster, sofern diese in den Daten vorhanden sind, zu zeigen.[3] In diesem Projekt deutete die Marchenko-Pastur-Verteilung darauf hin, dass die erste Hauptkomponente der alleinige Informationsträger war. Zur Absicherung der visuellen Ergebnisse wurden mehrere Clustering-Algorithmen durchgeführt (hierarchisches Clustering mittels Manhattan Distanz, k-Means und PAM Algorithmus sowie dbscan). Über den Einsatz zweier Parameter zur Bestimmung der Gruppenzuordnungsqualität eines jeden Patienten (z.B. der sogenannte Silhouetten-Breite) wurden dann die am besten differenzierten Cluster bestimmt. Durch das Markieren der so erhaltenen neuen Gruppierung der Patienten im PCA-Biplot konnte die neue Kategorisierung sichtbar gemacht und überprüft werden (siehe Abb. 2).
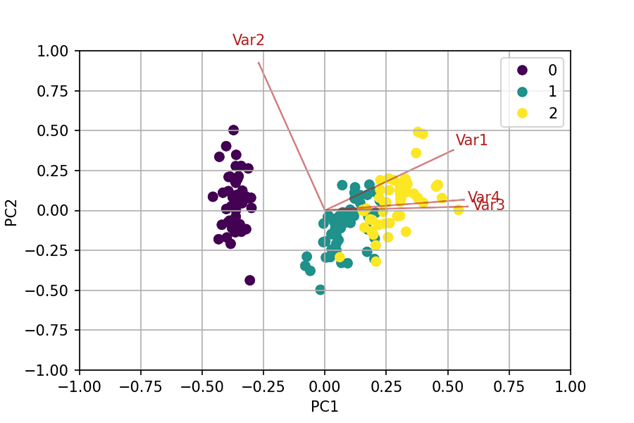
Eine bereits etablierte Kategorisierung, die ausschließlich auf Genexpression beruht, wurde mit der neuen Kategorisierung verglichen. Die Ergebnisse dieses Vergleichs wurden in einer Heatmap visualisiert. Die beiden Kategorisierungen konnten damit klar voneinander abgegrenzt werden. In dieser Studie dienten die Genexpressionsdaten des Tumors zusätzlich dazu, diejenigen biochemischen Prozesse zu identifizieren, die für die untersuchten Unterschiede im Gewebe verantwortlich sind. Die Ergebnisse aus dieser erfolgreichen Zusammenarbeit konnten in einem renommierten Wissenschaftsjournal zum Themengebiet Krebsforschung veröffentlicht werden. Sie werden die Entwicklung personalisierter Therapieansätze nachhaltig unterstützen.
Sie wollen auch mithilfe unseres Knowhows gezielt Information aus Ihren Daten holen? Dann zögern Sie nicht, uns zu kontaktieren!
Kontakt: Helen Schneider | Vertrieb helen.schneider@biovariance.com Quellen

