
The name mucoviscidosis, also called cystic fibrosis, comes from the Latin and means as much as sticky mucus. It‘s an autosomal recessive trait in which a mutation in the CFTR gene in gladular cells causes increased viscous and tough mucus leading to functional disorders in the affected organs.
Autosomal recessive inheritance
Diploid organisms have two copies of each gene – one from the mother and one from the father. A genome is homozygous when both copies (alleles) are identical and heterozygous when they differ. There is always one variant that determines the phenotyp. This allele is dominant over the other, which is recessive and doesn’t appear.
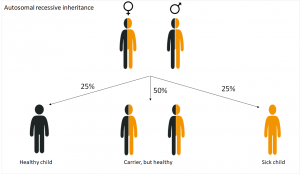
In diseases that are inherited in an autosomal recessive way, the genetic cause lays on one of the 22 autosomes . The parents carry the changed allele, which, however, doesn’t appear. So they are healthy.
A child suffers from cystic fibrosis when inherited the altered allele from both parents. This happens with a probability of 25%. Children who inherit both a wild-type and a sick allele are healthy, but carriers, like their parents. Although they don’t suffer from the disease, they can pass it on the next generation.
Ion channels
 In 1938, an American pediatrician described the disease for the first time and nearly 50 years later, the responsible gene was discovered:1 CFTR (cystic fibrosis transmembrane conductance regulator) consists of about 6,500 base pairs and is located on the long arm of chromosome 7.2
In 1938, an American pediatrician described the disease for the first time and nearly 50 years later, the responsible gene was discovered:1 CFTR (cystic fibrosis transmembrane conductance regulator) consists of about 6,500 base pairs and is located on the long arm of chromosome 7.2
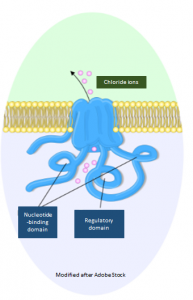
It contains a building instruction for a channel in the cell membrane through which ions are able to enter and leave the cell.3 Such channels, also called ion channels, are tubular protein complexes embedded in the membrane that are selective for certain ions.4 The gene CFTR encodes a chloride channel which occures mainly in epithelia of the intestine, pancreas, sweat glands and in the respiratory system. The 1,440 amico acid transmembrane protein consists of a regulatory subunit with multiple phosphorylation sites and two nucleotide-binding domains hydrolysing ATP to ADP.3 The main task of CFTR is the regulation of sodium – and water balance of human cells.3
Mutations in the CFTR gene
The CFTR proteins are the cause of the metabolic disorder. As a result, the ion transport is blocked and there is a disparity in the sodium – and water balance of the cells. These are no longer able to draw water into the surrounding tissue due to osmosis resulting in secretions with a too high salt content in lungs, sinuses, pancreas, intestine, bile duct and gonads.2
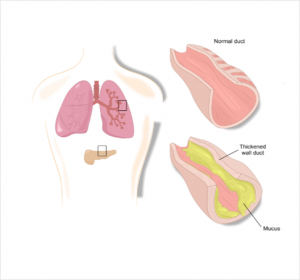 Due to the water removal, the water content in the corresponding secretions is too law – these become viscous and lead to funtional disorders in the affected organs.2 So far, almost 2,000 mutations are known, which are divided into six classes:5,6
Due to the water removal, the water content in the corresponding secretions is too law – these become viscous and lead to funtional disorders in the affected organs.2 So far, almost 2,000 mutations are known, which are divided into six classes:5,6
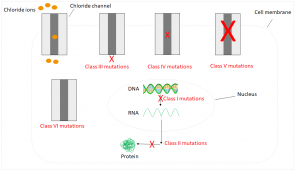
Class I mutations affect approximately 10% of all patients. They are caused by a defective mRNA, so the corresponding protein isn’t formed. In humans with class II mutations, the RNA is transcribed, but a mutation leads to a defective protein, which is subsequently enzymatically degraded. The most common case in Germany occuring in 70% of all patients is a deletion of three base pairs. The corresponding amino acid isn’t formed resulting in a functionless protein. Class III mutations lead to a regulatory disorder, so the channel isn’t open. Mutations of class IV leading to a reduced conductivity, which results in ions losing the ability to pass through the channel. Consequences of Class V and VI mutations are too few channels or unstable ones that are already degraded before reaching the cell membrane.5,6
Diseases
Cystic fibrosis is a multisystem disease due to the multiude of different mutations. The viscous mucus mainly clogs the fine branches of the bronchi and the excretory ducts of the internal organs, so the breathing and digestion are particularly affected.2
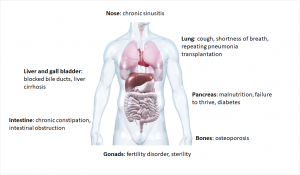
In the bronchi, the ion channel is primarily responsible for obtaining the liquid film on the surface by releasing the chloride ions. In this film small cilia move carrying dirt particles or bacteria out of the lungs. If this process is disrupted by increased mucus production, bacteria can more easily settle down and cause infections.7 Respiratory diseases are also the leading cause of death due to cystic fibrosis.1
In healthy people, the pancreas releases digestive enzymes into the duodenum. There the enzymes split proteins, carbohydrates and fats of food. In patients with cystic fibrosis this congests as viscous secretions. This leads to inflammation, the pancreas hardens and scarves (fibrosis). The consequences are malnutrition and growth disorders.2 The patients also have an increased risk of developing diabetes (Typ 3 diabetes: cystic fibrosis related diabetes, CRFD).8
Another consequence is infertility. Although 98% of men form fertilizable sperm, they can’t pass through the spermatic duct due to the mucus. Women are also only partially fertile, as the mucus in the fallopian tube doesn’t allow the sperm to penetrate.2,8
Common to all is the decreasing lung function, so often a transplantation is the last resort.1
Epidemiology
Cystic fibrosis is a rare disease with a prevalence of 1/3,300 – 1/4,800 newborns in Germany. Nevertheless, it is the second most common hereditary metabolic disease of the European population (after hemochromatosis). In Germany, there are currently 8,000 people affected, with about 200 new cases per year (as of 2017).5 Between 2012 and 2016, 423 people died from cystic fibrosis,9 in 2017 there were 48 cases of death.10 The frequency of heterozygote, so the proportion of all people carry a sick allele, is 4% in Europe (1:25).5
Current therapies
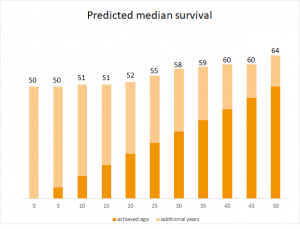
Even though the disease isn’t curable yet, life expectancy has increased in recent years.9 At that time children weren’t even one year old,1 now the predicted median survival of a newborn is 50 years.10 Currently, more than 50% of patients in Germany are older than 18 years.5 This is partly due to the fact that mutation-specific therapies are available for about one third of affected people.1 Genetic analyses detmine the exact cause of the disease. Since 2012, so-called CFTR modulators are available to improve or restore a defective or missing protein.1
In 2012 the European Commission approved the drug ivacaftor, which moderates the effects of the genetic defect.11 The CFTR potentiator is suitable for approximately 1,500 patients in Europe with class III mutations.12 Here, the protein is indeed in the cell membrane, but it doesn’t open. Ivacaftor restores this function and increases the transport of chloride ions through the channel. The basis for approval in Europa were the results of a phase III study in which patient‘ lung function was significantly improved.12 Lumacaftor is a member of the CFTR correctors and is used in class II mutations in combination with ivacaftor. The misfolded protein is is protected from intracellular enzymatic degradation and incorporated into the cell membrane. This year, another drug, tezacaftor, has been approved working in a similar way like lumacaftor and is also used in combination with ivacaftor.13 Studies have shown that the course of the disease was favorably influenced, for example by improving lung function.14,15
Another aspect is the implementation of so-called neonatal screenings. Two years ago, nationwide investigations of newborns on cystic fibrosis were introduced, which are for free and voluntary. In Bavaria, a total of 250,000 children were examinded in the period from September 2016 to August 2018. The congential disease was diagnosed in 45 children.16 This is important to start early therapy.
Outlook
Even though the metabolic disease isn’t curable yet, the chances due to CRISPR/Cas are relatively good. As a result, the pathogenic gene can be cut and replaced with a new one, or the existing mutation can be repaired.1
A recently discovered cell type could also be an approach for future therapies: Researchers have found a new typ in the respiratory mucosa and called it ionocyte. It appears to be the major producer of CFTR proteins and controls the expression of the chloride channels by its gene FOX1.17,18 Thus, regeneration of the ionocytes could lead to successful therapies.19,20 Once again, with these treatment options a progess is being made towards precision medicine. In total, 62 drugs have already been approved for personalized therapies.21
Contact Person:
Kristina Schraml (kristina.schraml@biovariance.com)

